
AADC deficiency from infancy to adulthood: Symptoms and developmental outcome in an international cohort of 63 patients
Pearson TS, et al. J Inherit Metab Dis. 2020.
Publication Date | May 2020
Authors | Pearson TS, Gilbert L, Opladen T, Garcia-Cazorla A, Mastrangelo M, Leuzzi V, Tay SKH, Sykut-Cegielska J, Pons R, Mercimek-Andrews S, Kato M, Lücke T, Oppebøen M, Kurian MA, Steel D, Manti F, Meeks KD, Jeltsch K, Flint L.
Citation | J Inherit Metab Dis. 2020;43(5):1121–30.
https://pubmed.ncbi.nlm.nih.gov/32369189/
A recent retrospective study,1 led by Toni Pearson at the Washington University School of Medicine, has highlighted the variety and complexity of motor and non-motor symptoms affecting patients with aromatic L-amino acid decarboxylase (AADC) deficiency throughout their lifespan.
AADC deficiency is a rare, autosomal recessive neurotransmitter disorder presenting early in life, in which a combined deficiency of serotonin, dopamine, norepinephrine and epinephrine leads to a complex syndrome characterised by motor, behavioural and autonomic symptoms.2 Most patients exhibit a severe disease course with profound motor impairment, with little or no benefit seen with existing medical interventions,2,3 while a smaller number of patients develop a milder disease2 and in general, respond more favourably to medical treatment.2,4–6 To further understand the evolution of symptoms throughout the disease course, the spectrum of developmental outcomes and mortality amongst patients with AADC deficiency, the international team of researchers analysed the responses to a questionnaire designed to assess the initial symptoms, symptom course, treatment response and developmental outcomes of patients with AADC deficiency.1
Patients with AADC deficiency from across the globe were identified from the International Working Group of Neurotransmitter-Related Disorders patient registry and the AADC Research Trust. Physicians and caregivers were asked to classify a range of symptoms as major, minor, or absent at different life stages from infancy to adulthood. The analysis included 63 patients from 23 countries (60% female; age range 6 months–37 years, median age 7 years; n=58 living).
The median age at the time of symptom onset was 3 months (range 0–12 months). There was considerable variation in the time from initial symptom onset to diagnosis of AADC deficiency, with younger patients being diagnosed more quickly than those aged over 10 years. The most common initial symptoms reported were hypotonia, oculogyric crises (OGCs), developmental delay, and feeding difficulties; consistent with previous reports.5 Prominent non-motor symptoms included sleepiness, irritability, excessive sweating, and nasal congestion (Figure 1).
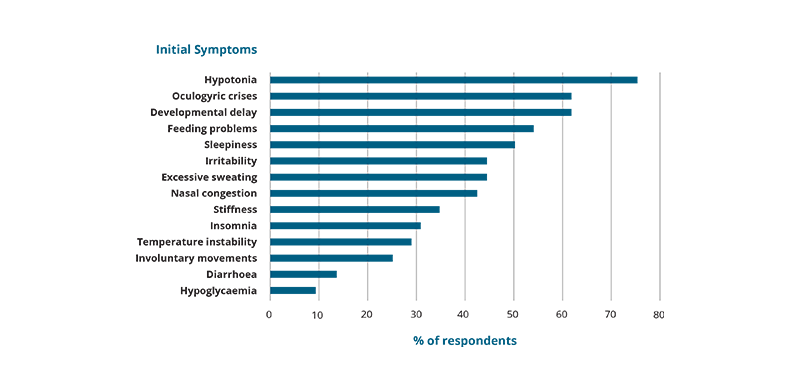
Adapted from: Pearson TS, et al. J Inherit Metab Dis. 2020;43:1121–30.
Figure 1: Prevalence of initial symptoms, as reported for 52 patients from the cohort.
Sleep disturbances appeared to evolve with age: excessive sleepiness was more prominent in children under the age of 2 years, whereas insomnia affected a large proportion of patients aged over 2 years and presented a major issue in 50% of children aged 2–12 years. Most children (85%) aged 6–12 years were also affected by irritability. Excessive sweating was also reported in all children under two years and classified as a major symptom in 40% of children between the ages of 2 and 17 years.
OGCs were reported by almost all patients but were more frequent, more severe, and longer lasting in younger patients (Figure 2). Episodes were particularly prevalent in those aged 2‒12 years (97%), comprising approximately half of the total study population. Of 5 older patients (≥13 years) who did not experience OGCs at the time of the survey, 4 had a milder disease and could walk independently, suggesting that the reduced prevalence of OGCs might be linked to either older age, reduced disease severity, or a combination of factors. An OGC was reported as the apparent proximate cause of death for 2 out of 5 deceased subjects, supporting the life-threatening potential of OGCs.
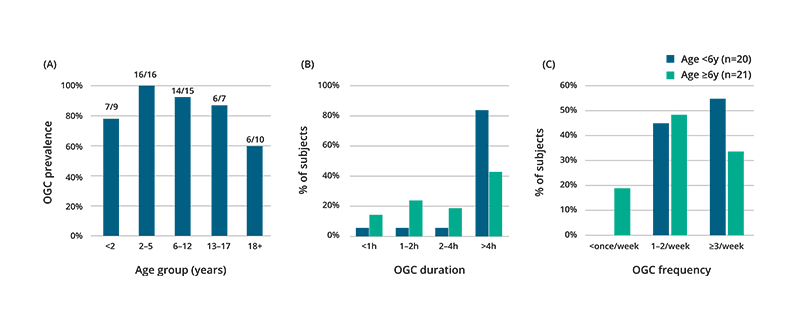
Adapted from: Pearson TS, et al. J Inherit Metab Dis. 2020;43:1121–30.
Figure 2: (A) Current prevalence of OGCs as reported for 57 patients (including 91% of subjects under 18 and 60% of subjects over 18); (B) OGC duration and (C) OGC frequency in younger (aged <6 years, blue) vs older (aged ≥6 years, green) patients.
49 patients aged 12 months or older reported information about the attainment of motor developmental milestones. Across these patients, only a third (16/49) exhibited head control, and only 22% (11/49) could sit and walk independently. Of those patients who could sit and walk independently, none were aged under 6 years. This reflects the notable difference in motor function across the age groups, with younger patients exhibiting more severe motor impairment than older patients. Indeed, most patients (7/11) who could walk were aged over 12 years. Investigators considered this to be indicative of a greater proportion of older patients in the cohort having a milder disease course, rather than reflective of a delay in the attainment of walking. Interestingly, early regression in motor or developmental skills was reported in 24% of 63 patients, which typically occurred during infancy at the onset of OCGs and other disease symptoms. Data from 53 patients showed that past or current feeding difficulties were experienced by 75% of patients. 45% of patients required a gastrostomy tube for feeding support.
Of the 38 subjects aged 5 years or older, only 4 (11%) were classed as completely independent, with 7 (18%) partially independent and 27 (71%) completely dependent. 62% (18/29) of those aged 5–18 years attended school and 2 of the 8 young adults aged over 18 years participated in work outside the home.
Of 43 subjects with a known genotype, 9 (21%) had a mild* motor phenotype, all of whom carried compound heterozygous variants including at least one missense variant. 14 novel variants identified were associated with a severe* phenotype in all but one subject.
Precise analysis of the childhood mortality risk was prohibited by the retrospective nature of the study. In addition, underreporting of some disease features due to partially incomplete responses for some subjects presented a further limitation of the study design.
The study illustrates the spectrum of disease phenotypes exhibited by patients with AADC deficiency throughout their lifespan. Whereas most patients experience severe functional and motor developmental impairment, others show milder symptoms. The authors note that the refractory nature of disease features to existing medical therapies, especially in severe cases, motivates the search for novel and effective therapeutic strategies.
- Pearson T, et al. J Inherit Metab Dis. 2020;43:1121–1130.
- Wassenberg T, et al. Orphanet J Rare Dis. 2017;12:12.
- Brun L, et al. Neurology. 2010;75:64–71.
- Leuzzi V, et al. JIMD Rep. 2015;15:39–45.
- Mastrangelo M, et al. Mov Disord Clin Pract. 2018;5:446–447.
- Tay SK, et al. Mol Genet Metab. 2007;91:374–378.